





PCR 是一個坑,坑里埋了好多研究僧……
PCR 是眾位「研究僧」日常實驗中常用的技術,可謂是「研究盛宴」中的一道「小菜」。可也有不少童鞋反映,這道菜不hao吃,一不小心就「小河里翻船」。
那么,我們該如何把這道菜「吃好」、跳出這個「坑」呢?
諸位看官莫急,待我送君「三大fa寶」。
一、確保 RNA 樣本合格
常見的 RNA 提取方法有 TRIzol 法、吸附法,這兩個方法各有所長,但都不能保證提取的 RNA 樣本一定合格。
因而,提取完成后應測定 RNA 的濃度及吸光度。通常,在 260,280,230 nm 處分別測定其吸光度(A260、A280、A230)并計算其比值(A260 /A280,A260 /A230)。

那么,這幾個數值及其比值分別有何意義?
A260 為核酸的吸光度,A280 為蛋白質的吸光度,A230 為其他雜質(多糖等)的吸光度。純 DNA 的 A260 /A280 為 1.8,純 RNA 的 A260 /A280 為 2.0。
如果 OD 比值不在范圍內該如何解決呢?
再次洗滌樣本!
75% 乙醇沉淀 RNA 后重新吸附、洗滌;若采用 TRIzol 法提取 RNA,可直接洗滌兩次。洗滌后離心時可兩次分別將 EP 管置于不同的方向,便于*洗滌 RNA 樣本。通常洗滌兩次后可使測得的 OD 值較為理想。
二、 利用「RNAstructure」軟件
引物,是進行熒光定量實驗的*條件。其特異性的問題可通過「BLAST」搞定,讓人比較頭痛的是引物可以形成「發夾結構」或「二聚體」(即在<75℃ 時出現溶解曲線的峰),影響擴增效率,終導致實驗結果不可用。
那么,這個問題能否解決呢?
有!設計引物的時候用「RNAstructure」預測一下引物的二級結構即可。

那么,這個軟件要如何使用呢?
1. 打開「RNAstructure」,單擊左上角的「new sequence」按鈕:
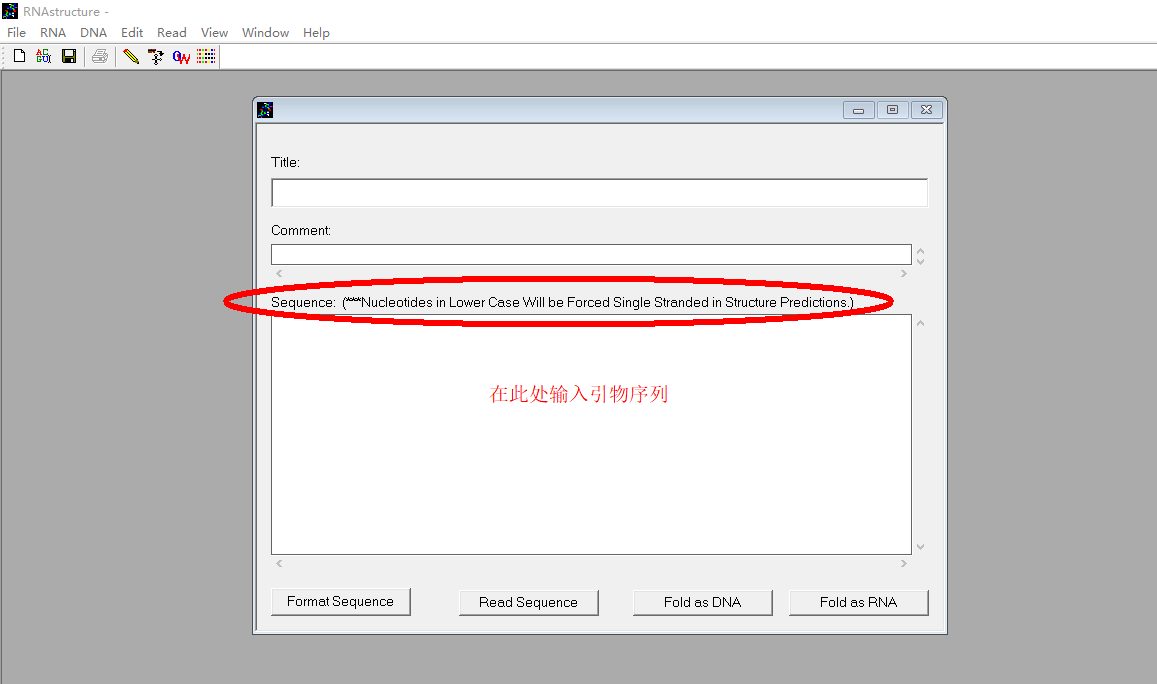
2. 新建序列文件,會出現一個對話框,輸入待檢測的引物序列:
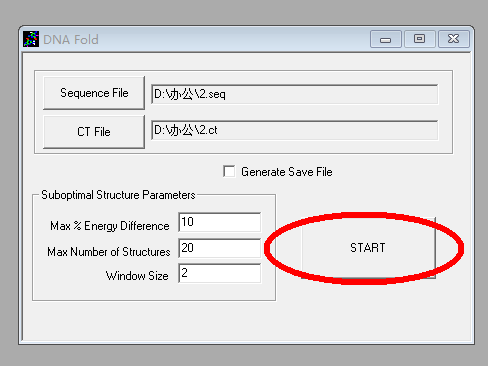
3. 單擊「Fold as DNA」,「save changes」,出現新的對話框后直接點擊「Start」:
4. 無需更改其他參數,再次點擊「Draw structure」,即可得到目標序列的二級結構:
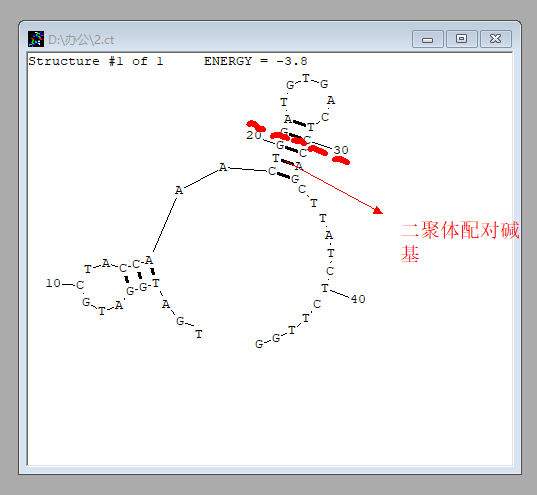
若考察引物能否形成發夾結構,則直接檢測 Forward 或 Reverse 引物即可。
若考察引物能否形成二聚體,則把兩條引物一起輸入,視作一條序列進行檢測即可。
在得到二級結構圖后,只要沒有連續 4 個以上的配對即可;若一條引物長度為 20 bp,在 18~22 處有連續 5 個的堿基配對,則可「手動拆解配對」,即保證引物為兩條鏈時的配對不多于 4 個即可。
三、保證擴增效率符合要求
有童鞋反映,兩個同類基因測試同一批樣本,理論上趨勢應該一致,結果卻是兩種趨勢,應該相信哪個呢?
不知道各位看官有沒有遇到類似的問題?這類問題該如何解決呢?
先說說出現這類問題的原因吧:
1. 擴增條件非*;
2. 樣本中含有抑制擴增的物質,如乙醇。
一對引物有其特定的 Tm 值,但 Tm 值時未必是其*擴增溫度。溫度過高時,擴增效率過低,差異部分的目的基因不能被擴增,導致「假陰性」結果的出現。
那么,如何確定擴增效率是否滿足要求呢?
通常,拿到新引物后應從 Tm- 5℃ 尋找其適宜的擴增溫度,在某溫度起為特異性擴增。
然后,在特異性擴增的溫度下檢測擴增效率:將 cDNA 梯度稀釋(一般為 2×),測試稀釋后的樣本 Ct 值差異是否為 1 左右。
若差值為 1,則擴增效率良好,可進行正式實驗。
若在特異性擴增的溫度范圍內不能使 Ct 值差異為 1,則需考慮重新設計引物。